
Abstract
Protein and lipid oxidation—mainly by mitochondrial reactive oxygen species (mtROS)—was proposed as a crucial determinant of health and lifespan. Angiotensin II (Ang II) enhances ROS production by activating NAD(P)H oxidase and uncoupling endothelial nitric oxide synthase (NOS). Ang II also stimulates mtROS production, which depresses mitochondrial energy metabolism. In rodents, renin–angiotensin system blockade (RAS blockade) increases survival and prevents age-associated changes. RAS blockade reduces mtROS and enhances mitochondrial content and function. This suggests that Ang II contributes to the ageing process by prompting mitochondrial dysfunction. Since Ang II is a pleiotropic peptide, the age-protecting effects of RAS blockade are expected to involve a variety of other mechanisms. Caloric restriction (CR)—an age-retarding intervention in humans and animals—and RAS blockade display a number of converging effects, i.e. they delay the manifestations of hypertension, diabetes, nephropathy, cardiovascular disease, and cancer; increase body temperature; reduce body weight, plasma glucose, insulin, and insulin-like growth factor-1; ameliorate insulin sensitivity; lower protein, lipid, and DNA oxidation, and mitochondrial H2O2 production; and increase uncoupling protein-2 and sirtuin expression. A number of these overlapping effects involve changes in mitochondrial function. In CR, peroxisome proliferator-activated receptors (PPARs) seem to contribute to age-retardation partly by regulating mitochondrial function. RAS inhibition up-regulates PPARs; therefore, it is feasible that PPAR modulation is pivotal for mitochondrial protection by RAS blockade during rodent ageing. Other potential mechanisms that may underlie RAS blockade’s mitochondrial benefits are TGF-β down-regulation and up-regulation of Klotho and sirtuins. In conclusion, the available data suggest that RAS blockade deserves further research efforts to establish its role as a potential tool to mitigate the growing problem of age-associated chronic disease.
1. The renin–angiotensin system and oxidants
The classical concept of the renin–angiotensin system (RAS) as a group of extracellular enzymes [renin, angiotensin-converting enzyme (ACE)] and circulating peptides [angiotensinogen, angiotensin I, angiotensin II (Ang II), and related peptides] that is exclusively involved in the regulation of systemic blood pressure and renal electrolyte balance—i.e. the circulating RAS—has evolved to incorporate the local or tissue RAS that comprises locally produced components serving as autocrine regulators of organ functions.1 RAS effects are mediated mainly by Ang II, which is responsible for vasoconstriction and Na+ retention.2 Ang II is also a pro-inflammatory and a pro-fibrotic agent.3 At present, it is clear that both the increased generation of cellular reactive oxygen species (ROS) and activation of redox-sensitive signalling cascades are critical events involved in Ang II actions.4 After binding to its AT1 receptors (AT1Rs), Ang II triggers intracellular superoxide (O2.−) production by activating NAD(P)H oxidase [NAD(P)H-ox]5,6 and uncoupling endothelial NOS (eNOS).7 Ang II also enhances nitric oxide (NO) generation8 and, since the reaction of NO with O2.− generates peroxynitrite, can promote the production of both ROS and reactive nitrogen species (RNS) and reduce NO availability.7,9 Under normal physiological conditions, Ang II-mediated ROS and RNS production, and the resulting stimulation of redox-sensitive signalling pathways are closely regulated.9 However, under conditions associated with RAS overactivation, such as hypertension, diabetes,10,11 and normal ageing,12–15 Ang II-dependent oxidant generation becomes a significant contributor to cell oxidation and tissue damage.16,17
2. Mitochondria as oxidant sources
Paralleling the evolution of scientific knowledge regarding the RAS, mitochondria—once viewed as essential organelles engaged exclusively in cellular energy production—are now recognized as key participants in the regulation of Ca2+ homeostasis,18 tissue O2 gradients,19 cell apoptosis,20 and intracellular signalling.21 However, as a result of partial O2 reduction by the respiratory chain, mitochondria are major cellular ROS sources and are themselves targets of ROS-mediated damage.22 A description of mitochondrial ROS (mtROS) sources and metabolism is out of the scope of this review; for an update, the reader may refer to other recent publications.23,24
3. The RAS–mitochondria interaction: role of ROS
In recent years, it became evident that RAS and mitochondria are engaged in mutual interactions through the production of ROS. Thus, Ang II not only activates NAD(P)H-ox and favours eNOS uncoupling, but also stimulates mtROS production, which in turn depresses mitochondrial energy metabolism.25,26 Ang II-mediated mtROS generation is triggered by the increase in cytosolic ROS levels that results from NAD(P)H-ox activation.26 Conversely, Ang II-induced and NAD(P)H-ox-mediated elevation of mtROS was found to increase the expression of NAD(P)H-ox subunits.27 Since NAD(P)H-ox activity is stimulated by H2O2 and lipid peroxides,28 it was suggested that mtROS may feed forwardly stimulate NAD(P)H-ox expression and activity.29
Importantly, the increase in mtROS production induced by Ang II is associated with vascular cell adhesion molecule-1 expression, a cytokine involved in atherosclerosis lesion formation,30 and decreased NO production in endothelial cells.26 Furthermore, recent evidence showed that mtROS contribute to in vivo Ang II-induced myocardial hypertrophy, sustained vascular dysfunction and ROS generation, and hypertension.27 Of note, in the latter study, overexpression of the mitochondrial antioxidant enzyme thioredoxin-2 protected against increases in vascular, myocardial, and mtROS, cardiovascular changes, and hypertension during chronic Ang II infusion in mice. Another link between Ang II and mitochondrial dysfunction is suggested by data showing that mitochondrial p66Shc plays a crucial role in Ang II-induced myocardial remodelling.31 p66Shc, partially localized in the mitochondrial intermembrane space, was suggested to contribute to mtROS production by subtracting electrons from cytochrome c and transferring them to oxygen to generate superoxide.
Finally, direct interactions between Ang II and nuclear and mitochondrial components have also been reported, including the existence of nuclear AT1-like Ang II receptors32,33 and detection of Ang II within nuclei and mitochondria.32,34–36
Summarizing, accumulating evidence indicates that mtROS contribute to the deleterious effects of Ang II, which may be mediated by activation of AT1Rs or by direct interaction of Ang II with mitochondrial or nuclear components.
4. ROS, mitochondria, and ageing
Oxidative damage to cellular macromolecules is thought to act as a driving force in the ageing process.37 The key contribution of mitochondria to continuous ROS and RNS production supports the mitochondrial free radical (FR) theory of ageing,38 as an extension of the more general FR theory of ageing.39
According to the mitochondrial version of the FR theory of ageing, ROS—mainly mtROS—oxidize mitochondrial components and alter mitochondrial function, which further enhances mtROS production. The resulting oxidative damage to proteins and lipids leads to cell and organ function deterioration and crucially determines health and lifespan.
Although substantial evidence indicates that oxidative damage augments during ageing, the question of whether mitochondrial oxidative stress causally contributes to ageing continues unsettled.40 At present, on the basis of studies in a variety of species, some researches seriously question the FR theory of ageing,41,42 whereas others continue to provide evidence in favour of the pivotal role played by mtROS in age-associated changes and survival.43,44
5. RAS blockade goes beyond blood pressure control: a strategy against ageing that protects mitochondria?
In the early 1990s, it became evident that in addition to its haemodynamic actions, Ang II acted as a growth factor45,46 and data had started to accumulate concerning its pro-fibrotic actions.47 These newly discovered actions of Ang II emerged from earlier work showing that RAS blockade with ACEinhibitors (ACEi) and later with AT1R blockers (ARB) in the setting of hypertension, heart failure, and chronic renal failure provided cardiac and renal benefits not limited to their antihypertensive effects (reviewed in Weber et al.48 and Kang et al.49). In this line, ACEi administration to experimental diabetic rats and renal ablated rats markedly reduced glomerulosclerosis.50,51 The progressive development of glomerulosclerosis is a well-known phenomenon of the ageing kidney.52 Since the issue of protection against age-associated glomerulosclerosis had not been previously addressed, we decided to evaluate the effects of ACEi on renal ageing. We found that enalapril, administered from weaning until 24 months of age, attenuates renal and glomerular age-related changes in CF1 mice.53 Later, we found that the above enalapril treatment also decreased myocardial sclerosis, increased the heart and liver mitochondrial number, prevented age-associated weight loss, and increased survival in ageing mice.54 These findings led us to believe that in enalapril-treated animals, the natural ageing mechanisms had been altered and that the RAS played a role in ageing. We hypothesized that enalapril had acted as an antioxidant thereby preventing mitochondrial injury. To the best of our knowledge, this was the first study to report that ACEi treatment increases rodent survival and protects mitochondria. The latter action on mice survival is in agreement with our recent data in rats.55
To further investigate the participation of RAS in the ageing process, we focused our research on the effects of RAS blockade on mitochondrial function. Since mitochondrial dysfunction and RAS have been independently involved in hypertension56 and diabetes,57 we hypothesized that RAS blockade might protect mitochondria from damage associated with both pathological conditions. In spontaneously hypertensive rats (SHR), losartan treatment prevented the alterations in renal mitochondrial H2O2 production rate, membrane potential, uncoupling protein (UCP)-2 content, and Mn-SOD, mitochondrial NOS, and cytochrome oxidase activities that occurred in untreated SHR.58 In diabetic rats by streptozotocin injection, losartan protected kidney mitochondria against changes in H2O2 production rate, membrane potential, and pyruvate content, without reducing plasma glucose levels.59 In both studies, treatment with amlodipine, a Ca2+ channel blocker, lowered blood pressure to a level similar to that of losartan treatment but showed no beneficial actions on kidney mitochondria alterations.
In agreement with our findings in the kidney, Ang II inhibition improved cardiac mitochondria energy production,60–62 and in diabetic rats, captopril treatment up-regulated the expression of energy production genes.63 Regarding the potential factor(s) that may mediate the effects of Ang II inhibitors on mitochondrial function, a study suggested that mitochondrial NO contributes to enalapril’s renal mitochondrial actions.64 Other reports showed that ARB can modulate UCP mRNA levels in the mouse brown adipose tissue65 and rat liver,66 or UCP content in the rat kidney.64,67 UCPs, by uncoupling mitochondrial electron transport from ATP production, can modify mitochondrial energy output, as well as decrease mitochondrial oxidant production. Evidence supporting a role for UCP-induced uncoupling in the attenuation of cell damage by excess ROS during ageing was very recently reviewed.68
In addition to the above-mentioned benefits of long-term ACEi or ARB treatments on cardiac and renal mitochondrial function during rodent ageing, other age-related protective actions include body fat mass reduction and improvements in physical performance69 and cognitive function.70
We also investigated the consequences of RAS blockade on age-associated mitochondrial DNA (mtDNA) alterations. In rats, enalapril or losartan administration for 16.5 months was unable to prevent the age-dependent accumulation of liver mtDNA ‘common deletion’, but attenuated the decrease in mtDNA content.71 Also, both treatments enhanced nuclear respiratory factor (NRF)-1 and peroxisome proliferator activator receptor-γ coactivator-1α (PGC-1α) mRNA contents. The above evidence seems to explain why enalapril and losartan improved mitochondrial functioning and lowered oxidant production,71 since both the absolute number of mtDNA molecules72 and increased NRF-1 and PGC-1 transcription are positively related to mitochondrial respiratory capacity;73,74 also, PGC-1 protects against the increases in ROS production and ROS-mediated damage.75
Recently, disruption of the AT1R gene was found to promote mice longevity, which was associated with protection against cardiac and vascular damage, oxidative damage in multiple organs, and with increases in the renal mitochondrial number and the expression of two pro-survival genes—nicotinamide phosphoribosyl-transferase (Nampt) and sirtuin 3 (Sirt3)—when compared with wild-type mice.76
5.1 Sirtuins, RAS, and ageing
Sirtuins are NAD+-dependent deacetylases that regulate the biological function of their targets by removing acetyl groups from acetyllysine-modified proteins. From unicellular organisms to mammals, sirtuins enhance organism and tissue survival in response to stress and toxicity.77 Mammalian SIRT7 seems to positively regulate RNA polymerase I transcription of ribosomal RNA genes and to be required for cell viability,78 and was proposed to improve tissue integrity in aged animals.79 Interestingly, SIRT1 overexpression modulates mitochondrial biogenesis by deacetylating PGC-1α.80 SIRT3—which is transported from nuclei to mitochondria upon cellular stress81—plays a role in mitochondrial functioning by deacetylating acetyl-CoA syntethase-282,83 and is the only sirtuin that seems to have a direct link to increased lifespan in humans.84 Sirt3 and Nampt (whose protein product increases mitochondrial NAD+, providing the co-substrate for SIRT3) are highly expressed in mitochondria, and their up-regulation contributes to increase cell survival during calorie restriction.81,85 In addition to SIRT1 and SIRT3, SIRT4 and SIRT5 were directly implicated in mitochondrial activity due to their localization to the mitochondria.86 Of note, overexpression of SIRT1 in vascular smooth muscle cells (VSMC), and resveratrol-induced activation of SIRT1in mice, down-regulates AT1R mRNA and protein levels, suggesting that the longevity and antiatherogenic effect of resveratrol is mediated at least partly by RAS blockade.87 Another link between sirtuins and RAS is apparent from results showing that in cultured murine tubular epithelial cells, Ang II down-regulates SirT3 mRNA and AT1R blockade (AT1R-bl) inhibits this effect,76 suggesting that targeting of Ang II/AT1R signalling may influence mammalian lifespan.88 Recently, SIRT6 was shown to attenuate NF-κB-dependent gene expression by deacetylating histone H3 lysine 9 (H3K9) at NF-κB target gene promoters. Hyperactivity of NF-κB signalling seems to contribute to premature and normal ageing.89 These results reveal a potential link between metabolism, inflammation, and ageing. Finally, in hamsters that had been administered resveratrol, nuclear SIRT1 induced mitochondrial Mn-SOD, which reduced oxidative stress and contributed to cardiomyocyte protection; also, by enhancing nuclear SIRT1 that in turn increased Mn-SOD levels, resveratrol suppressed myoblast death induced by Ang II.90
5.2 Klotho and RAS
Ang II infusion down-regulates renal klotho gene expression at both the mRNA and protein levels in an AT1R-dependent, but pressor-independent manner,91,92 and—after down-regulation by Ang II—losartan increases Klotho mRNA and protein expression in cultured tubular epithelial cells.93
The klotho gene, which encodes a single-pass transmembrane protein mainly expressed in kidney tubules, functions as an ageing-suppressor gene by abolishing the expression of diverse age-related phenotypes.91 In mice, klotho gene disruption accelerates the development of ageing-like phenotypes,94 whereas its overexpression extends lifespan.95,96 In a model of mouse glomerulonephritis, Klotho displayed a remarkable renoprotective effect associated with the improvement of mitochondrial function and mitigation of mitochondrial oxidative stress.97 Therefore, it is feasible that at least some of RAS blockade’s benefits in ageing are mediated through modulation of klotho expression.
5.3 Pleiotropic Ang II effects
Ang II is a pleiotropic peptide that by acting on AT1R and AT2 receptors in various tissues is involved in inflammation, cell growth, proliferation, immune response regulation,98 and central neuromodulation.99 In consequence, when analysing the beneficial effects of RAS blockade on ageing, it is necessary to consider an ample range of potential underlying mechanisms. In this line, long-term AT1R-bl, in addition to its cardio- and nephroprotective actions, prevents the increases of serum leptin, insulin, and glucose levels in ageing Fisher rats, indicating that metabolic control may contribute to the benefits of RAS blockade.100 In this context, in homozygous mice in which the ACE gene had been deleted, body fat was reduced independently of food intake—apparently as a result of increased hepatic fatty acid metabolism—and glucose tolerance was improved.101
In a recent review, we hypothesized that the Ang II-related depression of mitochondrial energy metabolism may result from the derangement of cytoskeletal and/or extracellular matrix (ECM) organization that is known to be induced by Ang II, and that TGF-β1 release is a potential link between Ang II, ECM, and cytoskeleton derangements, and mitochondrial dysfunction.102
The above data suggest that RAS activation contributes to the ageing process and supports the involvement of the Ang II–AT1R interaction as a relevant step leading to mitochondrial dysfunction.
Finally, an additional link between the RAS and ageing relates to the recently discovered AT1R-associated protein that reduces the number of AT1Rs at the surface of cardiovascular cells, decreases downstream Ang II signalling,103 and negatively regulates Ang II-induced senescence of VSMC, at least partly by inactivating the calcineurin/NFAT pathway.104 After being activated by calcineurin, NFAT translocates to the nucleus where it activates the transcription of genes encoding various molecules that participate in inflammation and Ca2+ dysregulation.
6. Converging effects of caloric restriction and RAS blockade: are peroxisome proliferator-activated receptors a common link?
Independently of the current controversy over the validity of the FR theory of ageing, there are at least two interventions that display age-retarding effects in humans and animals, i.e. caloric restriction (CR)105 and RAS blockade.54,55,76,88,106,107
Interestingly, both in humans and animal models, a number of physiological and pathological conditions are affected in a similar manner in response to RAS blockade or CR. As shown in Table 1, the converging effects of the latter interventions include: (i) delaying the manifestations of hypertension, diabetes, nephropathy, cardiovascular disease, and cancer; (ii) increasing body temperature and reducing body weight; (iii) lowering plasma insulin-like growth factor-1 (IGF-1); (iv) lowering of plasma glucose and insulin in hypertensive patients and rats; (v) amelioration of insulin sensitivity in hypertensive patients; (vi) lowering proteins, lipid, and DNA oxidation; (vii) diminution of mitochondrial H2O2 production rate accompanied by decreased mtDNA oxidation in rats and increased UCP-2 expression in mice and in humans; and (viii) up-regulation of sirtuins. Regarding the effects of RAS blockade and CR on weight loss, a positive correlation between plasma angiotensinogen levels and body mass index was reported in humans.108 However, the fact that Ang II can produce anorexia109 may afford an alternative interpretation for Ang II-mediated weight loss.
Table 1
Converging effects of CR and RAS blockade
| Effect | Species (reference) | |
|---|---|---|
| Calorie restriction | RAS blockade | |
| Retarding the manifestations of | ||
| Hypertension | Animal models,154 primates155 | Animal models,151 human150 |
| Diabetes | Primate,156 human157 | Human158 |
| Nephropathy | Rat159,160 | Human161 |
| Cardiovascular disease | Primate156 | Human162 |
| Cancer | Mice,163 primate156 | Rodent,164 human165,166 |
| Increasing body temperature | Rodent, primate167 | Rat168 |
| Loss of body weight | Primate169 | Rodent, human109 |
| Lowering plasma IGF-1 | Rodent, human170 | Rat,171 human172 |
| Reduction of plasma glucose and | ||
| insulin | Primate, rodent167 | Rat,173–175 human176 |
| Improvement of insulin sensitivity | Primate177 | Human178,179 |
| Reduction of | ||
| Protein oxidation | Mice,180,181 primate182 | Rat,183 human184 |
| Lipid oxidation | Rat,185 rat mitoch186,187 | Rat,188 human184 |
| DNA oxidation | Rat189–192 | Rat,193 human194 |
| Mitochondrial H2O2 production | Rat190,195 | Rat58,64,67,71 |
| Mitochondrial DNA damage | Rat190–192,195 | Rat71 |
| Increased expression of UCP-2 | Rodent,196 human197 | Rat64,67 |
| Up-regulation of sirtuins | Mouse liver cells129 | Mouse liver cells76 |
| Lifespan increase | Rodent,198 primate199 | Mice,54,76 rat55 |
IGF-1, insulin-like growth factor-1; UCP-2, uncoupling protein 2.
It should be noted that a number of the overlapping conditions that benefit from—or molecular events that occur in—experimental CR and experimental and clinical RAS blockade involve changes in mitochondrial functions, including hypertension;56,58 diabetes;57 the pathogenesis of cardiovascular damage,110 metabolic syndrome and obesity,111 diabetes mellitus (DM),59,112 renal disease,113 and atherosclerosis;114 insulin resistance;115 and the modulation of IGF-1 levels.116
In our search for a potential molecular link between CR and RAS blockade that could explain the above converging effects of these interventions, peroxisome proliferator-activated receptors (PPARs) emerged as an interesting possibility. PPARs are nuclear transcription factors that regulate the expression of a number of genes related to lipid metabolism and energy homeostasis in response to nutritional and physiological signals (such as exercise and cold),117 and the expression of genes involved in inflammatory processes.118 PPAR-α activation results in an increased expression of many nuclear genes associated with mitochondrial function, including those involved in fatty acid uptake, activation and β-oxidation,119 mitochondrial proton leak,120,121 and those encoding antioxidant enzymes, i.e. Mn-SOD and catalase.122 PPAR-γ, which is highly expressed in adipocytes, is involved in adipocyte differentiation and controls lipid storage gene expression. Another relevant function of PPAR-γ is the promotion of insulin sensitivity.123 PPAR-δ is involved in the regulation of fatty acid catabolism, metabolic rate, and mitochondrial proliferation mainly in skeletal muscles.124 Interaction with the retinoid X-receptor is necessary for PPAR-α, -γ, and -δ to exert their actions. In addition, induction of both PPAR-α and -γ target genes necessitates that these nuclear receptors interact with PGC-1, usually in complex with other coactivators and enzymes.125
PGC-1α and -1β also coactivate NRFs in those tissues that rely on aerobic metabolism, including the skeletal muscle, heart, and brown adipose tissue. NRFs activate the expression of genes involved in mitochondrial function and mtDNA content regulation and induce mitochondrial biogenesis.126,127 Noteworthily, the mechanisms that regulate PGC-1α and -1β functions include not only phosphorylation and methylation, but also acetylation by acetyl transferases—that inhibits their activities128—and deacetylation by SIRT1—that activates both cofactors.129,130
A very recent review describes the non-metabolic roles of PPARs, such as the regulation of tissue inflammation and hypertrophy, oxidative stress and ECM remodelling, the cell cycle, and angiogenesis.131
Ageing is associated with declines in PPAR-α and -γ expression132 and PPAR activities.133 Furthermore, PPARs seem to play an important role in the delay of ageing by dietary restriction.133 CR induces a generalized increase in PPAR activity, preventing their age-related decrease.132 Available evidence suggests that PPARs play a pivotal role in the association between CR and longevity, partly by reducing oxidative stress through modulation of UCP expression, improving forkhead box class O factor (FOXO) activity, and suppressing the transcription of pro-inflammatory NF-κB (for review, see Nunn et al.134). FOXO are a group of transcription factors involved in resistance to stress.
Interestingly, PPAR-γ regulates the expression of anti-ageing klotho.135 In this context, both the PPAR-γ agonist pioglitazone and increased renal expression of PPAR-γ protected from age-related renal damage, which was associated with increased klotho expression and with decreases in systemic and renal oxidative stress and mitochondrial injury.136
In this scenario, RAS blockade is associated with increased PPARs expression. Thus, enalapril up-regulates PPAR-α and -γ and displays antiatherogenic and anti-inflammatory effects in mice.137 Two ARB, irbesartan and telmisartan, were identified as PPAR-γ activators,138,139 whereas a product of losartan’s hepatic metabolism (EXP3179) was identified as a partial PPAR-γ agonist, suggesting that some ARB can mediate AT1R-independent effects.140
Recapitulating, the evidence provided above suggests that: (i) mitochondrial function and oxidant production are active participants in the ageing process; (ii) a number of the physiological benefits and molecular events that occur in experimental CR and experimental and clinical RAS blockade involve changes in mitochondrial function; (iii) PPARs, by regulating mitochondrial function and UCP, seem to play a major role in the age-retarding effects of CR; and (iv) RAS blockade delays the deleterious effects of ageing and also up-regulates PPARs. By integrating these findings, it is possible to hypothesize that PPAR modulation is a joint event that may underlie the mitochondrial protective actions associated with both CR and RAS blockade inhibition during rodent ageing.
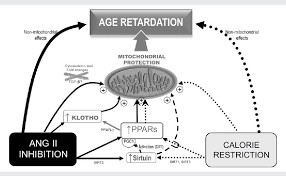
Figure 1 shows a schematic representation of several potential mechanisms that may contribute to the age-retarding effects associated with RAS blockade, where PPAR up-regulation serves as a common link between age-retardation by RAS blockade and CR. The mechanisms depicted here have a common feature: they all positively influence mitochondria. Considering that mitochondrial oxidative decay is thought to be central to the ageing process, it can be suggested that RAS blockade retards rodent ageing at least partly by protecting mitochondria.
Figure 1

The scheme shows several mechanisms that may contribute to the age-retarding effects associated with RAS blockade, i.e. up-regulation of PPARs, up-regulation of sirtuins and klotho gene expression, and cytoskeletal and ECM changes, all of which can have a stimulatory effect on mitochondrial function. Apart from other effects that are specific for each intervention, both CR and RAS blockade were shown to up-regulate PPARs and sirtuins. Overexpression of SIRT1 modulates mitochondrial biogenesis by deacetylating PGC-1α, and SIRT3 plays a role in mitochondrial functioning by deacetylating acetyl-CoA synthethase 2. RAS blockade up-regulates anti-ageing Klotho, and we hypothesize that by up-regulating PPARs CR may also up-regulate Klotho. Our hypothesis on a central participation of PPARs in the retardation of ageing mediated by RAS blockade is based on evidence showing that (i) mitochondrial function and oxidant production are active participants in the ageing process; (ii) RAS blockade delays the deleterious effects of ageing, improves mitochondrial function, and also up-regulates PPARs; (iii) PPARs, by regulating mitochondrial function and UCPs, seem to play a major role in the age-retarding effects of CR; and (iv) experimental CR and experimental and clinical RAS blockade display overlapping physiological and molecular events (Table 1), most of which involve changes in mitochondrial function. Future work will allow to either confirm or refute this hypothesis.
Recently, Cassis et al.88 reviewed evidence showing that after binding to its AT1R, Ang II participates in normal ageing by promoting oxidative damage to mitochondria and the ensuing mitochondrial dysfunction. In those studies, the detrimental age-related actions of Ang II were revealed first by registering the consequences of pharmacological Ang II blockade in ageing, and more recently were confirmed in mice that carried an experimentally disrupted AT1 subtype A receptor (AT1AR) gene. The authors also point to CR, administration of resveratrol (a plant-derived substance found in wine), lack of the IGF-1 receptor, and disruption of mouse AT1AR as a group of experimental interventions that prolong lifespan and—as a common feature—protect mitochondria and cell integrity through the modulation of ROS generation and sirtuin expression. They concluded that since ARB are modern and well-tolerated therapeutic agents for hypertension and cardiac failure, they might also constitute a strategy aimed at attenuating age-related disease and extending lifespan, notwithstanding that further studies are needed to validate animal results in humans. In this review, we also discussed experimental evidence supporting the benefits of Ang II blockade in the retardation of age-related changes, including the role of reduced oxidant generation, mitochondrial protection, and sirtuins. However, in attempt to identify additional molecular players in Ang II blockade’s age-retarding effects, we have changed the angle by analysing the interplay of Ang II blockade and CR in protecting mitochondria from oxidative stress, which ultimately results in prolonged lifespan. As a result of this analysis, we have emphasized the role of PPAR modulation as a joint event that may underlie the mitochondrial protective actions associated with both CR and RAS blockade during rodent ageing. We also commented on the modulation klotho gene expression (an age-suppressor gene) as a potential player in at least some of RAS blockade’s benefits in ageing. Finally, it should be mentioned that since heterozygous inactivation of the IGF-1 receptor in mice increases lifespan and resistance to oxidative stress,141 it is feasible that lowering of plasma IGF-1 participates in the beneficial effects of RAS blockade.
Most of the ageing studies reviewed here were conducted in rodents; therefore, their results cannot be directly translated to humans. Future research is needed to unravel whether RAS blockade protects against human ageing as well. Also, in several of our studies, to investigate the preventive actions of RAS blockade against age-related changes, ACEi or ARB were administered to healthy animals during a major part of their lifetime. Consequently, we cannot provide data relating to the potential reversion of incipient or already installed age-associated changes. However, other researchers showed that ARB (losartan) can induce the regression of age-related glomerular and vascular sclerosis in normotensive rats,142 and both ACEi (temocaprilat) and ARB (olmesartan) reverse advanced cardiac hypertrophy in ageing SHR.143 Also, evidence supporting the regression of renal lesions by RAS blockade in humans was reviewed by Remuzzi et al.144
Although it is out of the scope of this review, it is interesting to note that abundant information is available regarding both Ang II blockade’s benefits in diabetes and its functional, structural, and biochemical effects in the heart. In this regard, recent reviews have addressed the role of RAS blockers in retarding or preventing the onset and progression of DM,145 type 2 diabetes,146,147 and cardiovascular disease,147,148 and the potential mechanisms involved in the beneficial metabolic and cardiovascular actions of these compounds.145,146,148
In the context of the present controversial subject of combined ACEi/ARB therapy, experimental and clinical data have demonstrated that ACE inhibition initially decreases Ang II plasma levels; however, during prolonged treatment, plasmatic Ang II may rise up to initial values. This phenomenon—called ACE escape—possibly results from mast cell chymase-dependent conversion of Ang I into Ang II and increased ACE gene expression. Dual blockade of the RAS by the combined use of ACEi and ARB emerged as a rational solution to circumvent ACE escape that would prevent Ang II binding to AT1R, while permitting the stimulation of vacant AT2 receptors (which mediate effects mainly opposing those of AT1R activation). However, although there is no information available concerning combined ACEi/ARB therapy as either a preventive strategy or treatment against incipient vascular, cardiac, or renal lesions, the analysis of various studies conducted in patients with advanced vascular damage revealed that the combination of ACEi and ARB is associated with more adverse events than monotherapy, without exceeding the latter’s advantages.149
At present, it is not feasible to either evaluate the effects of RAS blockade as a preventive intervention against ageing in healthy individuals or analyse its cost/benefit relation. However, both animal and human evidence show that RAS blockade can prevent age-related structural and functional alterations in several organs, progression to the metabolic syndrome, the development of diabetes, hypertension and some of its consequences, cardiovascular changes, and cerebral and cognitive impairments.150–153 The latter conditions act as surrogate markers of the ageing process, and at the same time, they accelerate age-related structural and functional decay in various tissues.
In conclusion, the available data suggests that RAS blockade deserves further research efforts to establish its role as a potential tool to mitigate the growing problem of age-associated chronic disease.
Conflict of interest: none declared.
Abstract
Protein and lipid oxidation—mainly by mitochondrial reactive oxygen species (mtROS)—was proposed as a crucial determinant of health and lifespan. Angiotensin II (Ang II) enhances ROS production by activating NAD(P)H oxidase and uncoupling endothelial nitric oxide synthase (NOS). Ang II also stimulates mtROS production, which depresses mitochondrial energy metabolism. In rodents, renin–angiotensin system blockade (RAS blockade) increases survival and prevents age-associated changes. RAS blockade reduces mtROS and enhances mitochondrial content and function. This suggests that Ang II contributes to the ageing process by prompting mitochondrial dysfunction. Since Ang II is a pleiotropic peptide, the age-protecting effects of RAS blockade are expected to involve a variety of other mechanisms. Caloric restriction (CR)—an age-retarding intervention in humans and animals—and RAS blockade display a number of converging effects, i.e. they delay the manifestations of hypertension, diabetes, nephropathy, cardiovascular disease, and cancer; increase body temperature; reduce body weight, plasma glucose, insulin, and insulin-like growth factor-1; ameliorate insulin sensitivity; lower protein, lipid, and DNA oxidation, and mitochondrial H2O2 production; and increase uncoupling protein-2 and sirtuin expression. A number of these overlapping effects involve changes in mitochondrial function. In CR, peroxisome proliferator-activated receptors (PPARs) seem to contribute to age-retardation partly by regulating mitochondrial function. RAS inhibition up-regulates PPARs; therefore, it is feasible that PPAR modulation is pivotal for mitochondrial protection by RAS blockade during rodent ageing. Other potential mechanisms that may underlie RAS blockade’s mitochondrial benefits are TGF-β down-regulation and up-regulation of Klotho and sirtuins. In conclusion, the available data suggest that RAS blockade deserves further research efforts to establish its role as a potential tool to mitigate the growing problem of age-associated chronic disease.
1. The renin–angiotensin system and oxidants
The classical concept of the renin–angiotensin system (RAS) as a group of extracellular enzymes [renin, angiotensin-converting enzyme (ACE)] and circulating peptides [angiotensinogen, angiotensin I, angiotensin II (Ang II), and related peptides] that is exclusively involved in the regulation of systemic blood pressure and renal electrolyte balance—i.e. the circulating RAS—has evolved to incorporate the local or tissue RAS that comprises locally produced components serving as autocrine regulators of organ functions.1 RAS effects are mediated mainly by Ang II, which is responsible for vasoconstriction and Na+ retention.2 Ang II is also a pro-inflammatory and a pro-fibrotic agent.3 At present, it is clear that both the increased generation of cellular reactive oxygen species (ROS) and activation of redox-sensitive signalling cascades are critical events involved in Ang II actions.4 After binding to its AT1 receptors (AT1Rs), Ang II triggers intracellular superoxide (O2.−) production by activating NAD(P)H oxidase [NAD(P)H-ox]5,6 and uncoupling endothelial NOS (eNOS).7 Ang II also enhances nitric oxide (NO) generation8 and, since the reaction of NO with O2.− generates peroxynitrite, can promote the production of both ROS and reactive nitrogen species (RNS) and reduce NO availability.7,9 Under normal physiological conditions, Ang II-mediated ROS and RNS production, and the resulting stimulation of redox-sensitive signalling pathways are closely regulated.9 However, under conditions associated with RAS overactivation, such as hypertension, diabetes,10,11 and normal ageing,12–15 Ang II-dependent oxidant generation becomes a significant contributor to cell oxidation and tissue damage.16,17
2. Mitochondria as oxidant sources
Paralleling the evolution of scientific knowledge regarding the RAS, mitochondria—once viewed as essential organelles engaged exclusively in cellular energy production—are now recognized as key participants in the regulation of Ca2+ homeostasis,18 tissue O2 gradients,19 cell apoptosis,20 and intracellular signalling.21 However, as a result of partial O2 reduction by the respiratory chain, mitochondria are major cellular ROS sources and are themselves targets of ROS-mediated damage.22 A description of mitochondrial ROS (mtROS) sources and metabolism is out of the scope of this review; for an update, the reader may refer to other recent publications.23,24
3. The RAS–mitochondria interaction: role of ROS
In recent years, it became evident that RAS and mitochondria are engaged in mutual interactions through the production of ROS. Thus, Ang II not only activates NAD(P)H-ox and favours eNOS uncoupling, but also stimulates mtROS production, which in turn depresses mitochondrial energy metabolism.25,26 Ang II-mediated mtROS generation is triggered by the increase in cytosolic ROS levels that results from NAD(P)H-ox activation.26 Conversely, Ang II-induced and NAD(P)H-ox-mediated elevation of mtROS was found to increase the expression of NAD(P)H-ox subunits.27 Since NAD(P)H-ox activity is stimulated by H2O2 and lipid peroxides,28 it was suggested that mtROS may feed forwardly stimulate NAD(P)H-ox expression and activity.29
Importantly, the increase in mtROS production induced by Ang II is associated with vascular cell adhesion molecule-1 expression, a cytokine involved in atherosclerosis lesion formation,30 and decreased NO production in endothelial cells.26 Furthermore, recent evidence showed that mtROS contribute to in vivo Ang II-induced myocardial hypertrophy, sustained vascular dysfunction and ROS generation, and hypertension.27 Of note, in the latter study, overexpression of the mitochondrial antioxidant enzyme thioredoxin-2 protected against increases in vascular, myocardial, and mtROS, cardiovascular changes, and hypertension during chronic Ang II infusion in mice. Another link between Ang II and mitochondrial dysfunction is suggested by data showing that mitochondrial p66Shc plays a crucial role in Ang II-induced myocardial remodelling.31 p66Shc, partially localized in the mitochondrial intermembrane space, was suggested to contribute to mtROS production by subtracting electrons from cytochrome c and transferring them to oxygen to generate superoxide.
Finally, direct interactions between Ang II and nuclear and mitochondrial components have also been reported, including the existence of nuclear AT1-like Ang II receptors32,33 and detection of Ang II within nuclei and mitochondria.32,34–36
Summarizing, accumulating evidence indicates that mtROS contribute to the deleterious effects of Ang II, which may be mediated by activation of AT1Rs or by direct interaction of Ang II with mitochondrial or nuclear components.
4. ROS, mitochondria, and ageing
Oxidative damage to cellular macromolecules is thought to act as a driving force in the ageing process.37 The key contribution of mitochondria to continuous ROS and RNS production supports the mitochondrial free radical (FR) theory of ageing,38 as an extension of the more general FR theory of ageing.39
According to the mitochondrial version of the FR theory of ageing, ROS—mainly mtROS—oxidize mitochondrial components and alter mitochondrial function, which further enhances mtROS production. The resulting oxidative damage to proteins and lipids leads to cell and organ function deterioration and crucially determines health and lifespan.
Although substantial evidence indicates that oxidative damage augments during ageing, the question of whether mitochondrial oxidative stress causally contributes to ageing continues unsettled.40 At present, on the basis of studies in a variety of species, some researches seriously question the FR theory of ageing,41,42 whereas others continue to provide evidence in favour of the pivotal role played by mtROS in age-associated changes and survival.43,44
5. RAS blockade goes beyond blood pressure control: a strategy against ageing that protects mitochondria?
In the early 1990s, it became evident that in addition to its haemodynamic actions, Ang II acted as a growth factor45,46 and data had started to accumulate concerning its pro-fibrotic actions.47 These newly discovered actions of Ang II emerged from earlier work showing that RAS blockade with ACEinhibitors (ACEi) and later with AT1R blockers (ARB) in the setting of hypertension, heart failure, and chronic renal failure provided cardiac and renal benefits not limited to their antihypertensive effects (reviewed in Weber et al.48 and Kang et al.49). In this line, ACEi administration to experimental diabetic rats and renal ablated rats markedly reduced glomerulosclerosis.50,51 The progressive development of glomerulosclerosis is a well-known phenomenon of the ageing kidney.52 Since the issue of protection against age-associated glomerulosclerosis had not been previously addressed, we decided to evaluate the effects of ACEi on renal ageing. We found that enalapril, administered from weaning until 24 months of age, attenuates renal and glomerular age-related changes in CF1 mice.53 Later, we found that the above enalapril treatment also decreased myocardial sclerosis, increased the heart and liver mitochondrial number, prevented age-associated weight loss, and increased survival in ageing mice.54 These findings led us to believe that in enalapril-treated animals, the natural ageing mechanisms had been altered and that the RAS played a role in ageing. We hypothesized that enalapril had acted as an antioxidant thereby preventing mitochondrial injury. To the best of our knowledge, this was the first study to report that ACEi treatment increases rodent survival and protects mitochondria. The latter action on mice survival is in agreement with our recent data in rats.55
To further investigate the participation of RAS in the ageing process, we focused our research on the effects of RAS blockade on mitochondrial function. Since mitochondrial dysfunction and RAS have been independently involved in hypertension56 and diabetes,57 we hypothesized that RAS blockade might protect mitochondria from damage associated with both pathological conditions. In spontaneously hypertensive rats (SHR), losartan treatment prevented the alterations in renal mitochondrial H2O2 production rate, membrane potential, uncoupling protein (UCP)-2 content, and Mn-SOD, mitochondrial NOS, and cytochrome oxidase activities that occurred in untreated SHR.58 In diabetic rats by streptozotocin injection, losartan protected kidney mitochondria against changes in H2O2 production rate, membrane potential, and pyruvate content, without reducing plasma glucose levels.59 In both studies, treatment with amlodipine, a Ca2+ channel blocker, lowered blood pressure to a level similar to that of losartan treatment but showed no beneficial actions on kidney mitochondria alterations.
In agreement with our findings in the kidney, Ang II inhibition improved cardiac mitochondria energy production,60–62 and in diabetic rats, captopril treatment up-regulated the expression of energy production genes.63 Regarding the potential factor(s) that may mediate the effects of Ang II inhibitors on mitochondrial function, a study suggested that mitochondrial NO contributes to enalapril’s renal mitochondrial actions.64 Other reports showed that ARB can modulate UCP mRNA levels in the mouse brown adipose tissue65 and rat liver,66 or UCP content in the rat kidney.64,67 UCPs, by uncoupling mitochondrial electron transport from ATP production, can modify mitochondrial energy output, as well as decrease mitochondrial oxidant production. Evidence supporting a role for UCP-induced uncoupling in the attenuation of cell damage by excess ROS during ageing was very recently reviewed.68
In addition to the above-mentioned benefits of long-term ACEi or ARB treatments on cardiac and renal mitochondrial function during rodent ageing, other age-related protective actions include body fat mass reduction and improvements in physical performance69 and cognitive function.70
We also investigated the consequences of RAS blockade on age-associated mitochondrial DNA (mtDNA) alterations. In rats, enalapril or losartan administration for 16.5 months was unable to prevent the age-dependent accumulation of liver mtDNA ‘common deletion’, but attenuated the decrease in mtDNA content.71 Also, both treatments enhanced nuclear respiratory factor (NRF)-1 and peroxisome proliferator activator receptor-γ coactivator-1α (PGC-1α) mRNA contents. The above evidence seems to explain why enalapril and losartan improved mitochondrial functioning and lowered oxidant production,71 since both the absolute number of mtDNA molecules72 and increased NRF-1 and PGC-1 transcription are positively related to mitochondrial respiratory capacity;73,74 also, PGC-1 protects against the increases in ROS production and ROS-mediated damage.75
Recently, disruption of the AT1R gene was found to promote mice longevity, which was associated with protection against cardiac and vascular damage, oxidative damage in multiple organs, and with increases in the renal mitochondrial number and the expression of two pro-survival genes—nicotinamide phosphoribosyl-transferase (Nampt) and sirtuin 3 (Sirt3)—when compared with wild-type mice.76
5.1 Sirtuins, RAS, and ageing
Sirtuins are NAD+-dependent deacetylases that regulate the biological function of their targets by removing acetyl groups from acetyllysine-modified proteins. From unicellular organisms to mammals, sirtuins enhance organism and tissue survival in response to stress and toxicity.77 Mammalian SIRT7 seems to positively regulate RNA polymerase I transcription of ribosomal RNA genes and to be required for cell viability,78 and was proposed to improve tissue integrity in aged animals.79 Interestingly, SIRT1 overexpression modulates mitochondrial biogenesis by deacetylating PGC-1α.80 SIRT3—which is transported from nuclei to mitochondria upon cellular stress81—plays a role in mitochondrial functioning by deacetylating acetyl-CoA syntethase-282,83 and is the only sirtuin that seems to have a direct link to increased lifespan in humans.84 Sirt3 and Nampt (whose protein product increases mitochondrial NAD+, providing the co-substrate for SIRT3) are highly expressed in mitochondria, and their up-regulation contributes to increase cell survival during calorie restriction.81,85 In addition to SIRT1 and SIRT3, SIRT4 and SIRT5 were directly implicated in mitochondrial activity due to their localization to the mitochondria.86 Of note, overexpression of SIRT1 in vascular smooth muscle cells (VSMC), and resveratrol-induced activation of SIRT1in mice, down-regulates AT1R mRNA and protein levels, suggesting that the longevity and antiatherogenic effect of resveratrol is mediated at least partly by RAS blockade.87 Another link between sirtuins and RAS is apparent from results showing that in cultured murine tubular epithelial cells, Ang II down-regulates SirT3 mRNA and AT1R blockade (AT1R-bl) inhibits this effect,76 suggesting that targeting of Ang II/AT1R signalling may influence mammalian lifespan.88 Recently, SIRT6 was shown to attenuate NF-κB-dependent gene expression by deacetylating histone H3 lysine 9 (H3K9) at NF-κB target gene promoters. Hyperactivity of NF-κB signalling seems to contribute to premature and normal ageing.89 These results reveal a potential link between metabolism, inflammation, and ageing. Finally, in hamsters that had been administered resveratrol, nuclear SIRT1 induced mitochondrial Mn-SOD, which reduced oxidative stress and contributed to cardiomyocyte protection; also, by enhancing nuclear SIRT1 that in turn increased Mn-SOD levels, resveratrol suppressed myoblast death induced by Ang II.90
5.2 Klotho and RAS
Ang II infusion down-regulates renal klotho gene expression at both the mRNA and protein levels in an AT1R-dependent, but pressor-independent manner,91,92 and—after down-regulation by Ang II—losartan increases Klotho mRNA and protein expression in cultured tubular epithelial cells.93
The klotho gene, which encodes a single-pass transmembrane protein mainly expressed in kidney tubules, functions as an ageing-suppressor gene by abolishing the expression of diverse age-related phenotypes.91 In mice, klotho gene disruption accelerates the development of ageing-like phenotypes,94 whereas its overexpression extends lifespan.95,96 In a model of mouse glomerulonephritis, Klotho displayed a remarkable renoprotective effect associated with the improvement of mitochondrial function and mitigation of mitochondrial oxidative stress.97 Therefore, it is feasible that at least some of RAS blockade’s benefits in ageing are mediated through modulation of klotho expression.
5.3 Pleiotropic Ang II effects
Ang II is a pleiotropic peptide that by acting on AT1R and AT2 receptors in various tissues is involved in inflammation, cell growth, proliferation, immune response regulation,98 and central neuromodulation.99 In consequence, when analysing the beneficial effects of RAS blockade on ageing, it is necessary to consider an ample range of potential underlying mechanisms. In this line, long-term AT1R-bl, in addition to its cardio- and nephroprotective actions, prevents the increases of serum leptin, insulin, and glucose levels in ageing Fisher rats, indicating that metabolic control may contribute to the benefits of RAS blockade.100 In this context, in homozygous mice in which the ACE gene had been deleted, body fat was reduced independently of food intake—apparently as a result of increased hepatic fatty acid metabolism—and glucose tolerance was improved.101
In a recent review, we hypothesized that the Ang II-related depression of mitochondrial energy metabolism may result from the derangement of cytoskeletal and/or extracellular matrix (ECM) organization that is known to be induced by Ang II, and that TGF-β1 release is a potential link between Ang II, ECM, and cytoskeleton derangements, and mitochondrial dysfunction.102
The above data suggest that RAS activation contributes to the ageing process and supports the involvement of the Ang II–AT1R interaction as a relevant step leading to mitochondrial dysfunction.
Finally, an additional link between the RAS and ageing relates to the recently discovered AT1R-associated protein that reduces the number of AT1Rs at the surface of cardiovascular cells, decreases downstream Ang II signalling,103 and negatively regulates Ang II-induced senescence of VSMC, at least partly by inactivating the calcineurin/NFAT pathway.104 After being activated by calcineurin, NFAT translocates to the nucleus where it activates the transcription of genes encoding various molecules that participate in inflammation and Ca2+ dysregulation.
6. Converging effects of caloric restriction and RAS blockade: are peroxisome proliferator-activated receptors a common link?
Independently of the current controversy over the validity of the FR theory of ageing, there are at least two interventions that display age-retarding effects in humans and animals, i.e. caloric restriction (CR)105 and RAS blockade.54,55,76,88,106,107
Interestingly, both in humans and animal models, a number of physiological and pathological conditions are affected in a similar manner in response to RAS blockade or CR. As shown in Table 1, the converging effects of the latter interventions include: (i) delaying the manifestations of hypertension, diabetes, nephropathy, cardiovascular disease, and cancer; (ii) increasing body temperature and reducing body weight; (iii) lowering plasma insulin-like growth factor-1 (IGF-1); (iv) lowering of plasma glucose and insulin in hypertensive patients and rats; (v) amelioration of insulin sensitivity in hypertensive patients; (vi) lowering proteins, lipid, and DNA oxidation; (vii) diminution of mitochondrial H2O2 production rate accompanied by decreased mtDNA oxidation in rats and increased UCP-2 expression in mice and in humans; and (viii) up-regulation of sirtuins. Regarding the effects of RAS blockade and CR on weight loss, a positive correlation between plasma angiotensinogen levels and body mass index was reported in humans.108 However, the fact that Ang II can produce anorexia109 may afford an alternative interpretation for Ang II-mediated weight loss.
Table 1
Converging effects of CR and RAS blockade
| Effect | Species (reference) | |
|---|---|---|
| Calorie restriction | RAS blockade | |
| Retarding the manifestations of | ||
| Hypertension | Animal models,154 primates155 | Animal models,151 human150 |
| Diabetes | Primate,156 human157 | Human158 |
| Nephropathy | Rat159,160 | Human161 |
| Cardiovascular disease | Primate156 | Human162 |
| Cancer | Mice,163 primate156 | Rodent,164 human165,166 |
| Increasing body temperature | Rodent, primate167 | Rat168 |
| Loss of body weight | Primate169 | Rodent, human109 |
| Lowering plasma IGF-1 | Rodent, human170 | Rat,171 human172 |
| Reduction of plasma glucose and | ||
| insulin | Primate, rodent167 | Rat,173–175 human176 |
| Improvement of insulin sensitivity | Primate177 | Human178,179 |
| Reduction of | ||
| Protein oxidation | Mice,180,181 primate182 | Rat,183 human184 |
| Lipid oxidation | Rat,185 rat mitoch186,187 | Rat,188 human184 |
| DNA oxidation | Rat189–192 | Rat,193 human194 |
| Mitochondrial H2O2 production | Rat190,195 | Rat58,64,67,71 |
| Mitochondrial DNA damage | Rat190–192,195 | Rat71 |
| Increased expression of UCP-2 | Rodent,196 human197 | Rat64,67 |
| Up-regulation of sirtuins | Mouse liver cells129 | Mouse liver cells76 |
| Lifespan increase | Rodent,198 primate199 | Mice,54,76 rat55 |
IGF-1, insulin-like growth factor-1; UCP-2, uncoupling protein 2.
It should be noted that a number of the overlapping conditions that benefit from—or molecular events that occur in—experimental CR and experimental and clinical RAS blockade involve changes in mitochondrial functions, including hypertension;56,58 diabetes;57 the pathogenesis of cardiovascular damage,110 metabolic syndrome and obesity,111 diabetes mellitus (DM),59,112 renal disease,113 and atherosclerosis;114 insulin resistance;115 and the modulation of IGF-1 levels.116
In our search for a potential molecular link between CR and RAS blockade that could explain the above converging effects of these interventions, peroxisome proliferator-activated receptors (PPARs) emerged as an interesting possibility. PPARs are nuclear transcription factors that regulate the expression of a number of genes related to lipid metabolism and energy homeostasis in response to nutritional and physiological signals (such as exercise and cold),117 and the expression of genes involved in inflammatory processes.118 PPAR-α activation results in an increased expression of many nuclear genes associated with mitochondrial function, including those involved in fatty acid uptake, activation and β-oxidation,119 mitochondrial proton leak,120,121 and those encoding antioxidant enzymes, i.e. Mn-SOD and catalase.122 PPAR-γ, which is highly expressed in adipocytes, is involved in adipocyte differentiation and controls lipid storage gene expression. Another relevant function of PPAR-γ is the promotion of insulin sensitivity.123 PPAR-δ is involved in the regulation of fatty acid catabolism, metabolic rate, and mitochondrial proliferation mainly in skeletal muscles.124 Interaction with the retinoid X-receptor is necessary for PPAR-α, -γ, and -δ to exert their actions. In addition, induction of both PPAR-α and -γ target genes necessitates that these nuclear receptors interact with PGC-1, usually in complex with other coactivators and enzymes.125
PGC-1α and -1β also coactivate NRFs in those tissues that rely on aerobic metabolism, including the skeletal muscle, heart, and brown adipose tissue. NRFs activate the expression of genes involved in mitochondrial function and mtDNA content regulation and induce mitochondrial biogenesis.126,127 Noteworthily, the mechanisms that regulate PGC-1α and -1β functions include not only phosphorylation and methylation, but also acetylation by acetyl transferases—that inhibits their activities128—and deacetylation by SIRT1—that activates both cofactors.129,130
A very recent review describes the non-metabolic roles of PPARs, such as the regulation of tissue inflammation and hypertrophy, oxidative stress and ECM remodelling, the cell cycle, and angiogenesis.131
Ageing is associated with declines in PPAR-α and -γ expression132 and PPAR activities.133 Furthermore, PPARs seem to play an important role in the delay of ageing by dietary restriction.133 CR induces a generalized increase in PPAR activity, preventing their age-related decrease.132 Available evidence suggests that PPARs play a pivotal role in the association between CR and longevity, partly by reducing oxidative stress through modulation of UCP expression, improving forkhead box class O factor (FOXO) activity, and suppressing the transcription of pro-inflammatory NF-κB (for review, see Nunn et al.134). FOXO are a group of transcription factors involved in resistance to stress.
Interestingly, PPAR-γ regulates the expression of anti-ageing klotho.135 In this context, both the PPAR-γ agonist pioglitazone and increased renal expression of PPAR-γ protected from age-related renal damage, which was associated with increased klotho expression and with decreases in systemic and renal oxidative stress and mitochondrial injury.136
In this scenario, RAS blockade is associated with increased PPARs expression. Thus, enalapril up-regulates PPAR-α and -γ and displays antiatherogenic and anti-inflammatory effects in mice.137 Two ARB, irbesartan and telmisartan, were identified as PPAR-γ activators,138,139 whereas a product of losartan’s hepatic metabolism (EXP3179) was identified as a partial PPAR-γ agonist, suggesting that some ARB can mediate AT1R-independent effects.140
Recapitulating, the evidence provided above suggests that: (i) mitochondrial function and oxidant production are active participants in the ageing process; (ii) a number of the physiological benefits and molecular events that occur in experimental CR and experimental and clinical RAS blockade involve changes in mitochondrial function; (iii) PPARs, by regulating mitochondrial function and UCP, seem to play a major role in the age-retarding effects of CR; and (iv) RAS blockade delays the deleterious effects of ageing and also up-regulates PPARs. By integrating these findings, it is possible to hypothesize that PPAR modulation is a joint event that may underlie the mitochondrial protective actions associated with both CR and RAS blockade inhibition during rodent ageing.
Figure 1 shows a schematic representation of several potential mechanisms that may contribute to the age-retarding effects associated with RAS blockade, where PPAR up-regulation serves as a common link between age-retardation by RAS blockade and CR. The mechanisms depicted here have a common feature: they all positively influence mitochondria. Considering that mitochondrial oxidative decay is thought to be central to the ageing process, it can be suggested that RAS blockade retards rodent ageing at least partly by protecting mitochondria.
Figure 1
The scheme shows several mechanisms that may contribute to the age-retarding effects associated with RAS blockade, i.e. up-regulation of PPARs, up-regulation of sirtuins and klotho gene expression, and cytoskeletal and ECM changes, all of which can have a stimulatory effect on mitochondrial function. Apart from other effects that are specific for each intervention, both CR and RAS blockade were shown to up-regulate PPARs and sirtuins. Overexpression of SIRT1 modulates mitochondrial biogenesis by deacetylating PGC-1α, and SIRT3 plays a role in mitochondrial functioning by deacetylating acetyl-CoA synthethase 2. RAS blockade up-regulates anti-ageing Klotho, and we hypothesize that by up-regulating PPARs CR may also up-regulate Klotho. Our hypothesis on a central participation of PPARs in the retardation of ageing mediated by RAS blockade is based on evidence showing that (i) mitochondrial function and oxidant production are active participants in the ageing process; (ii) RAS blockade delays the deleterious effects of ageing, improves mitochondrial function, and also up-regulates PPARs; (iii) PPARs, by regulating mitochondrial function and UCPs, seem to play a major role in the age-retarding effects of CR; and (iv) experimental CR and experimental and clinical RAS blockade display overlapping physiological and molecular events (Table 1), most of which involve changes in mitochondrial function. Future work will allow to either confirm or refute this hypothesis.
Recently, Cassis et al.88 reviewed evidence showing that after binding to its AT1R, Ang II participates in normal ageing by promoting oxidative damage to mitochondria and the ensuing mitochondrial dysfunction. In those studies, the detrimental age-related actions of Ang II were revealed first by registering the consequences of pharmacological Ang II blockade in ageing, and more recently were confirmed in mice that carried an experimentally disrupted AT1 subtype A receptor (AT1AR) gene. The authors also point to CR, administration of resveratrol (a plant-derived substance found in wine), lack of the IGF-1 receptor, and disruption of mouse AT1AR as a group of experimental interventions that prolong lifespan and—as a common feature—protect mitochondria and cell integrity through the modulation of ROS generation and sirtuin expression. They concluded that since ARB are modern and well-tolerated therapeutic agents for hypertension and cardiac failure, they might also constitute a strategy aimed at attenuating age-related disease and extending lifespan, notwithstanding that further studies are needed to validate animal results in humans. In this review, we also discussed experimental evidence supporting the benefits of Ang II blockade in the retardation of age-related changes, including the role of reduced oxidant generation, mitochondrial protection, and sirtuins. However, in attempt to identify additional molecular players in Ang II blockade’s age-retarding effects, we have changed the angle by analysing the interplay of Ang II blockade and CR in protecting mitochondria from oxidative stress, which ultimately results in prolonged lifespan. As a result of this analysis, we have emphasized the role of PPAR modulation as a joint event that may underlie the mitochondrial protective actions associated with both CR and RAS blockade during rodent ageing. We also commented on the modulation klotho gene expression (an age-suppressor gene) as a potential player in at least some of RAS blockade’s benefits in ageing. Finally, it should be mentioned that since heterozygous inactivation of the IGF-1 receptor in mice increases lifespan and resistance to oxidative stress,141 it is feasible that lowering of plasma IGF-1 participates in the beneficial effects of RAS blockade.
Most of the ageing studies reviewed here were conducted in rodents; therefore, their results cannot be directly translated to humans. Future research is needed to unravel whether RAS blockade protects against human ageing as well. Also, in several of our studies, to investigate the preventive actions of RAS blockade against age-related changes, ACEi or ARB were administered to healthy animals during a major part of their lifetime. Consequently, we cannot provide data relating to the potential reversion of incipient or already installed age-associated changes. However, other researchers showed that ARB (losartan) can induce the regression of age-related glomerular and vascular sclerosis in normotensive rats,142 and both ACEi (temocaprilat) and ARB (olmesartan) reverse advanced cardiac hypertrophy in ageing SHR.143 Also, evidence supporting the regression of renal lesions by RAS blockade in humans was reviewed by Remuzzi et al.144
Although it is out of the scope of this review, it is interesting to note that abundant information is available regarding both Ang II blockade’s benefits in diabetes and its functional, structural, and biochemical effects in the heart. In this regard, recent reviews have addressed the role of RAS blockers in retarding or preventing the onset and progression of DM,145 type 2 diabetes,146,147 and cardiovascular disease,147,148 and the potential mechanisms involved in the beneficial metabolic and cardiovascular actions of these compounds.145,146,148
In the context of the present controversial subject of combined ACEi/ARB therapy, experimental and clinical data have demonstrated that ACE inhibition initially decreases Ang II plasma levels; however, during prolonged treatment, plasmatic Ang II may rise up to initial values. This phenomenon—called ACE escape—possibly results from mast cell chymase-dependent conversion of Ang I into Ang II and increased ACE gene expression. Dual blockade of the RAS by the combined use of ACEi and ARB emerged as a rational solution to circumvent ACE escape that would prevent Ang II binding to AT1R, while permitting the stimulation of vacant AT2 receptors (which mediate effects mainly opposing those of AT1R activation). However, although there is no information available concerning combined ACEi/ARB therapy as either a preventive strategy or treatment against incipient vascular, cardiac, or renal lesions, the analysis of various studies conducted in patients with advanced vascular damage revealed that the combination of ACEi and ARB is associated with more adverse events than monotherapy, without exceeding the latter’s advantages.149
At present, it is not feasible to either evaluate the effects of RAS blockade as a preventive intervention against ageing in healthy individuals or analyse its cost/benefit relation. However, both animal and human evidence show that RAS blockade can prevent age-related structural and functional alterations in several organs, progression to the metabolic syndrome, the development of diabetes, hypertension and some of its consequences, cardiovascular changes, and cerebral and cognitive impairments.150–153 The latter conditions act as surrogate markers of the ageing process, and at the same time, they accelerate age-related structural and functional decay in various tissues.
In conclusion, the available data suggests that RAS blockade deserves further research efforts to establish its role as a potential tool to mitigate the growing problem of age-associated chronic disease.
Conflict of interest: none declared.




{kind=link}