
Metabolic syndrome drives many of the hallmarks of aging, and multiple hallmarks of aging drive metabolic syndrome in a vicious, accelerating cycle.
Central obesity, high triglycerides, low HDL cholesterol, high blood pressure, and elevated fasting glucose comprise a cluster of metabolic disorders that increase the risk of cardiovascular disease and type 2 diabetes and define metabolic syndrome [1].
All of the factors of metabolic syndrome are associated not just with age but with accelerated aging. For example, age-associated inflammation can lead to insulin resistance, impaired glucose metabolism, and dyslipidemia, all of which are components of metabolic syndrome [2].
A healthy lifestyle can reverse or significantly slow the progress of metabolic syndrome in many instances [3, 4]. There are multiple mechanisms linking metabolic syndrome and the primary hallmarks of aging: genomic instability, telomere attrition, epigenetic alterations, altered proteostasis, and disabled macroautophagy [5].
Genomic instability
Metabolic syndrome is linked to an increased risk of genomic instability and thus DNA damage. This, in turn, increases the risk of cancer and other diseases [6, 7].
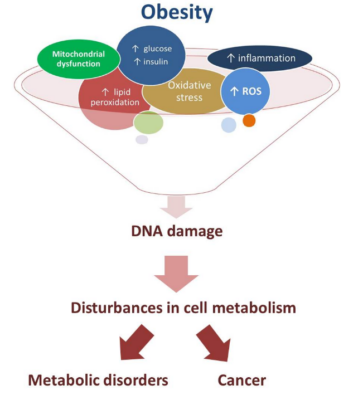
Factors associated with metabolic syndrome, including insulin resistance, oxidative stress, and chronic inflammation, drive genomic instability. Insulin resistance, for example, can cause increased production of insulin and insulin-like growth factors, which can promote the growth and proliferation of cells and increase the risk of DNA damage [8].
Oxidative stress, which occurs when there is an imbalance between the production of reactive oxygen species (ROS) and the body’s antioxidant defenses, can also contribute to genomic instability. ROS can damage DNA and other cellular components, leading to mutations and other forms of genomic instability [7].
Chronic inflammation, which is a hallmark of metabolic syndrome, can also contribute to genomic instability through the release of cytokines and other molecules that can damage DNA and increase the risk of mutation [9]. Furthermore, oxidative damage is also a product of inflammation [10].
Telomere attrition
Telomeres are the protective caps at the end of chromosomes. They play a critical role in maintaining genomic stability. Telomere attrition, which refers to the gradual shortening of telomeres over time, is a hallmark of aging [11].
Metabolic syndrome has been linked to accelerated telomere attrition through increased oxidative stress, inflammation, and insulin resistance. Oxidative stress can damage telomeric DNA, leading to telomere shortening [12], while inflammation can accelerate telomere attrition by promoting cellular aging and DNA damage. Insulin resistance, which is a hallmark of metabolic syndrome, has also been linked to telomere attrition, likely due to the role of insulin in promoting cell proliferation and DNA replication [13-15].
Epigenetic alterations
Epigenetic alterations are changes in the way that genes are expressed without changing the underlying DNA sequence. These alterations can occur in response to environmental factors such as diet, physical activity, and exposure to toxins. Several studies have suggested that epigenetic alterations play a role in metabolic syndrome and that metabolic syndrome in a vicious cycle drives further alterations in the epigenome.
Additionally, epigenetic changes have been associated with obesity, a key component of metabolic syndrome. Adipose tissue, which is the main place that the body stores fat, has been shown to undergo significant epigenetic changes in response to changes in nutrient availability and metabolic stress. These changes can contribute to insulin resistance and other metabolic abnormalities [16, 17]. For example, high-fat diets have been shown to induce changes in DNA methylation patterns that lead to altered gene expression in a way that favors metabolic syndrome [18].
Loss of proteostasis
Proteostasis refers to the maintenance of protein homeostasis, which is essential for cellular and organismal health. The loss of proteostasis, which can occur due to genetic and environmental factors, is associated with several age-related diseases, including metabolic syndrome [19].
Several mechanisms have been proposed to explain the relationship between the loss of proteostasis and metabolic syndrome. For example, the accumulation of misfolded or aggregated proteins can activate stress response pathways, such as the unfolded protein response (UPR), which can lead to insulin resistance and other metabolic abnormalities [20].
Furthermore, proteostasis is closely linked to mitochondrial function, which is essential for cellular energy metabolism. Mitochondrial dysfunction, which can result from impaired proteostasis, can contribute to metabolic abnormalities by reducing cellular energy production and increasing oxidative stress [21].
Finally, the loss of proteostasis can also contribute to chronic inflammation, which is a hallmark of metabolic syndrome. Misfolded or damaged proteins can activate the immune system and induce the release of inflammatory cytokines, which can contribute to insulin resistance and other metabolic abnormalities [22]. Conversely, metabolic syndrome can aggravate faltering proteostasis through several mechanisms.
Metabolic syndrome often leads to increased oxidative stress, which can damage proteins and make them misfold. This misfolding disrupts proteostasis and can lead to the formation of protein aggregates, which are harmful to cells [23].
Chronic inflammation, a common symptom of metabolic syndrome, can also disrupt proteostasis. Inflammation can lead to the production of cytokines that interfere with the normal functioning of the proteostasis network. For example, inflammation can affect the function of the endoplasmic reticulum, a cellular organelle involved in protein folding [24].
Insulin resistance can also disrupt proteostasis. Insulin signaling is crucial for many aspects of cellular function, including protein synthesis. Therefore, insulin resistance can lead to the overproduction or underproduction of certain proteins, disrupting the balance of the proteostasis network [25].
High levels of lipids, a common characteristic of metabolic syndrome, can cause lipotoxicity, leading to the impairment of various cellular processes, including proteostasis. High lipid levels can induce endoplasmic reticulum stress and initiate the unfolded protein response pathway, which can ultimately disrupt the balance of protein synthesis, folding, and degradation [26].
Lastly, prolonged high blood sugar levels can lead to the formation of advanced glycation end-products (AGEs). These AGEs can alter protein structure and function, thus contributing to impaired proteostasis [27].
Disabled autophagy
Autophagy is a process by which cells break down and recycle damaged or dysfunctional cellular components. Autophagy is essential for maintaining cellular homeostasis and is involved in a wide range of physiological processes, including nutrient metabolism, inflammation, and cellular stress response [28].
Disabled autophagy has been implicated in the development of metabolic syndrome. Several studies have shown that impaired autophagy is associated with insulin resistance, dyslipidemia, and other metabolic abnormalities [28, 29].
One potential mechanism by which impaired autophagy contributes to metabolic syndrome is through the accumulation of damaged organelles, such as mitochondria, which can lead to increased oxidative stress and inflammation [30].
Furthermore, autophagy plays a crucial role in the regulation of lipid metabolism. Impaired autophagy has been shown to result in the accumulation of lipids in cells and tissues, which can lead to dyslipidemia and non-alcoholic fatty liver disease (NAFLD), a common complication of metabolic syndrome [29].
Finally, autophagy is involved in the regulation of energy metabolism and glucose homeostasis. Impaired autophagy can lead to the accumulation of misfolded proteins and other cellular components, which can activate stress response pathways, such as the UPR, and lead to insulin resistance and impaired glucose metabolism [22].
Antagonistic and integrative hallmarks of aging
The integrative and antagonistic hallmarks of aging, which are interconnected and influence each other, are also affected by metabolic syndrome. They collectively contribute to the aging phenotype and age-related diseases. They include deregulated nutrient sensing, mitochondrial dysfunction, cellular senescence, stem cell exhaustion, altered intercellular communication, chronic inflammation and dysbiosis [31].
Deregulated nutrient sensing
Nutrient sensing is the ability of cells to sense and respond to changes in nutrient availability. This process plays a crucial role in the regulation of energy metabolism and is essential for maintaining metabolic homeostasis. Deregulated nutrient sensing, which can occur due to genetic and environmental factors, may lead to metabolic syndrome [32].
One way in which deregulated nutrient sensing can contribute to metabolic syndrome is through insulin resistance [33]. Insulin is a hormone that regulates glucose and lipid metabolism. Deregulated nutrient sensing can disrupt insulin signaling pathways, leading to impaired glucose and lipid metabolism along with insulin resistance [34].
Furthermore, deregulated nutrient sensing can lead to the accumulation of excess nutrients, such as glucose and fatty acids, in cells and tissues [35]. This can promote lipotoxicity, a condition in which excess lipids accumulate in tissues and lead to cellular dysfunction and inflammation. Lipotoxicity can contribute to insulin resistance and other metabolic abnormalities [36, 37].
Additionally, deregulated nutrient sensing can lead to chronic low-grade inflammation (inflammaging) [38], which is a hallmark of metabolic syndrome. Excess nutrient availability can activate immune cells and promote cytokine release [39].
Finally, deregulated nutrient sensing can contribute to NAFLD. Excess nutrient availability can lead to the accumulation of lipids in liver cells, leading to hepatic steatosis and NAFLD [40].
Mitochondrial dysfunction
Mitochondria are organelles within cells that play a critical role in energy production and metabolism. Mitochondrial dysfunction, which refers to impaired mitochondrial function, has been implicated in metabolic syndrome [41].
Mitochondrial dysfunction can contribute to metabolic syndrome through several mechanisms. For example, impaired mitochondrial function can lead to reduced energy production, which can impair glucose and lipid metabolism and contribute to insulin resistance and other metabolic abnormalities [42]. Mitochondrial dysfunction can also lead to the accumulation of ROS, can contribute to NAFLD [43], and can contribute to inflammation through immune cell and cytokine activation [44].
Cellular senescence
Cellular senescence is a process in which cells stop dividing and enter a state of irreversible growth arrest. Cellular senescence plays a crucial role in aging and is associated with several age-related diseases, including metabolic syndrome [45]. The inflammaging associated with metabolic syndrome also increases the risk of cellular senescence [46].
Furthermore, metabolic syndrome is associated with oxidative stress, which can contribute to cellular senescence by inducing DNA damage and other forms of cellular stress. Oxidative stress can activate stress response pathways, such as the p53 pathway, which can induce cellular senescence [47]. Telomere attrition can also contribute to cellular senescence by activating stress response pathways and inducing cellular damage [48].
Finally, metabolic syndrome is associated with impaired proteostasis, which can contribute to cellular senescence by inducing the accumulation of misfolded or damaged proteins, leading to the activation of stress response pathways and other forms of cellular stress [49].
Stem cell exhaustion
Stem cell exhaustion refers to the depletion of the pool of stem cells in tissues due to aging or other factors. Stem cells play a crucial role in tissue regeneration and repair, and their exhaustion has been implicated in the development of several age-related diseases, including metabolic syndrome [41].
Metabolic syndrome is associated with the accumulation of cellular damage and stress, which can contribute to the depletion of stem cells. Inflammaging, oxidative stress, and impaired proteostasis can all induce cellular damage and stress and contribute to stem cell exhaustion [50].
Furthermore, metabolic syndrome is associated with impaired tissue regeneration and repair, which can be attributed, at least in part, to the depletion of stem cells. Impaired tissue regeneration and repair can lead to the accumulation of damage and stress, contributing to metabolic abnormalities [50, 51].
In addition, metabolic syndrome has been associated with impaired angiogenesis, which is the process by which new blood vessels form from pre-existing ones. Angiogenesis is essential for tissue regeneration and repair, and its impairment can alsocontribute to metabolic abnormalities [52].
Finally, metabolic syndrome is associated with insulin resistance, which can impair the function of stem cells through reactive oxygen species production, activation of inflammatory pathways, and disruption of the insulin/IGF signaling pathway. Insulin resistance can contribute to the depletion of stem cells and impair tissue regeneration and repair [34, 53, 54].
Altered intercellular communication and metabolic syndrome
Intercellular communication refers to the exchange of information between cells, which is essential for maintaining cellular homeostasis and physiological function. Alterations in intercellular communication have been implicated in the development of metabolic syndrome [41], and the inflammaging associated with metabolic syndrome can lead to intercellular communication disruption [55, 56].
Furthermore, altered intercellular communication can impair the function of adipose tissue, which plays a crucial role in energy metabolism and is dysregulated in metabolic syndrome. Adipose tissue communicates with other tissues through the release of adipokines, which can influence glucose and lipid metabolism [57]. Alterations in adipokine secretion can contribute to insulin resistance and other metabolic abnormalities [58].
In addition, altered intercellular communication can impair the function of the gut microbiota, which plays a critical role in nutrient metabolism and the regulation of inflammation. The gut microbiota communicates with host cells through the release of metabolites and other molecules, which can influence energy metabolism and immune function. Alterations in the gut microbiota can contribute to insulin resistance and other metabolic abnormalities [59]. Finally, altered intercellular communication can contribute to NAFLD as well [60, 61].
Inflammaging
Inflammaging, which is a common link between metabolic syndrome and other hallmarks of aging, is associated with insulin resistance, dyslipidemia, and hypertension [62]. Several factors can contribute to this, including excessive nutrients leading to immune activation [63]. Metabolic stress, such as that caused by insulin resistance, can also activate immune cells and promote inflammation [64].
Another factor that can contribute to chronic inflammation in metabolic syndrome is the accumulation of visceral adipose tissue. Adipose tissue produces pro-inflammatory cytokines, such as tumor necrosis factor-alpha (TNF-alpha) and interleukin-6 (IL-6), which can contribute to insulin resistance and other metabolic abnormalities [65].
Furthermore, chronic inflammation can lead to the activation of stress response pathways, such as the UPR [66]. Chronic inflammation can also contribute to NAFLD [60].
Dysbiosis
Dysbiosis refers to an imbalance in the gut microbiota, the complex community of microorganisms that inhabit the gastrointestinal tract. Dysbiosis has been implicated in the development of several diseases, including metabolic syndrome [67].
The gut microbiota plays a critical role in nutrient metabolism and the regulation of inflammation. Dysbiosis can impair the function of the gut microbiota and contribute to metabolic abnormalities [67].
One of these pathways is insulin resistance [68, 69]. Dysbiosis can lead to the production of lipopolysaccharides (LPS), a molecule found in the outer membrane of certain bacteria, which can induce low-grade inflammation and contribute to insulin resistance [70]. Dysbiosis is linked to inflammaging [71] and NAFLD [72] as well.
Finally, dysbiosis can contribute to obesity, a key component of metabolic syndrome. Dysbiosis can alter the production of hormones and other molecules that regulate appetite and energy metabolism, leading to increased caloric intake and decreased energy expenditure [68].
Current and emerging strategies for managing metabolic syndrome
Given the multifaceted nature of metabolic syndrome and its association with the hallmarks of aging, the steps to mitigate its development should also be multi-targeted. There are potential steps that may influence metabolic syndrome and its effects on the hallmarks.
Regular physical activity and a balanced diet rich in fruits, vegetables, lean proteins, and whole grains can help maintain metabolic homeostasis, regulate nutrient sensing, and reduce excess nutrient accumulation in tissues. This can also help reduce obesity [73-75].
Anti-inflammatory diets that are rich in antioxidants and omega-3 fatty acids, or anti-inflammatory medications may help mitigate inflammaging [76]. Metformin and other glucose-lowering drugs can be beneficial in managing deregulated nutrient sensing and improving insulin sensitivity when used under medical supervision [77].
Supplements and interventions that support mitochondrial health, such as Coenzyme Q10 and PQQ, can potentially offset the mitochondrial dysfunction associated with metabolic syndrome [78]. Drugs like senolytics, which selectively remove senescent cells, could be helpful in managing metabolic syndrome [79].
Probiotics, prebiotics, and a diet promoting a healthy gut microbiome can address dysbiosis and reduce its contribution to metabolic syndrome [80]. Although this is a newer area of research, stem cell therapy could potentially help replenish the exhausted stem cell pool and support tissue regeneration and repair [81].
In conclusion, metabolic syndrome significantly intersects with the hallmarks of aging, and studies suggest that metabolic syndrome can accelerate aging by exacerbating these hallmarks.
Effective management and prevention of metabolic syndrome, therefore, could serve as a strategy for promoting healthy aging and delaying the onset of age-related diseases. This underscores the importance of early intervention and the adoption of healthy lifestyles, including a balanced diet and regular physical activity. Future research should continue exploring these critical interactions, ultimately leading to improved therapeutic approaches that address the multifaceted nature of aging and metabolic health.
Literature
[1] A. Tchernof and J. P. Després, “Pathophysiology of human visceral obesity: an update,” Physiol Rev, vol. 93, no. 1, pp. 359–404, Jan. 2013
[2] F. Bonomini, L. F. Rodella, and R. Rezzani, “Metabolic syndrome, aging and involvement of oxidative stress,” Aging Dis, vol. 6, no. 2, pp. 109–120, 2015
[3] N. J. Stone, “Successful control of dyslipidemia in patients with metabolic syndrome: focus on lifestyle changes,” Clin Cornerstone, vol. 8, no. SUPPL. 1, 2006
[4] P. M. Nilsson, J. Tuomilehto, and L. Rydén, “The metabolic syndrome – What is it and how should it be managed?,” Eur J Prev Cardiol, vol. 26, no. 2_suppl, pp. 33–46, Dec. 2019
[5] C. López-Otín, M. A. Blasco, L. Partridge, M. Serrano, and G. Kroemer, “Hallmarks of aging: An expanding universe,” Cell, vol. 186, no. 2, pp. 243–278, Jan. 2023
[6] S. Furukawa et al., “Increased oxidative stress in obesity and its impact on metabolic syndrome,” J Clin Invest, vol. 114, no. 12, pp. 1752–1761, May 2017
[7] M. Wlodarczyk and G. Nowicka, “Obesity, DNA Damage, and Development of Obesity-Related Diseases,” International Journal of Molecular Sciences 2019, Vol. 20, Page 1146, vol. 20, no. 5, p. 1146, Mar. 2019
[8] M. S. Lewitt, M. S. Dent, and K. Hall, “The Insulin-Like Growth Factor System in Obesity, Insulin Resistance and Type 2 Diabetes Mellitus,” Journal of Clinical Medicine 2014, Vol. 3, Pages 1561-1574, vol. 3, no. 4, pp. 1561–1574, Dec. 2014
[9] L. M. Coussens and Z. Werb, “Inflammation and cancer,” Nature 2002 420:6917, vol. 420, no. 6917, pp. 860–867, Dec. 2002
[10] M. Mittal, M. R. Siddiqui, K. Tran, S. P. Reddy, and A. B. Malik, “Reactive oxygen species in inflammation and tissue injury,” Antioxid Redox Signal, vol. 20, no. 7, pp. 1126–1167, Mar. 2014
[11] C. López-Otín, M. A. Blasco, L. Partridge, M. Serrano, and G. Kroemer, “The hallmarks of aging,” Cell, vol. 153, no. 6, p. 1194, Jun. 2013
[12] R. P. Barnes, E. Fouquerel, and P. L. Opresko, “The impact of oxidative DNA damage and stress on telomere homeostasis,” Mech Ageing Dev, vol. 177, pp. 37–45, Jan. 2019
[13] D. Révész et al., “Associations between cellular aging markers and metabolic syndrome: Findings from the cardia study,” Journal of Clinical Endocrinology and Metabolism, vol. 103, no. 1, pp. 148–157, 2018
[14] D. Révész, Y. Milaneschi, J. E. Verhoeven, and B. W. J. H. Penninx, “Telomere length as a marker of cellular aging is associated with prevalence and progression of metabolic syndrome,” Journal of Clinical Endocrinology and Metabolism, vol. 99, no. 12, pp. 4607–4615, Dec. 2014
[15] D. Révész, Y. Milaneschi, J. E. Verhoeven, J. Lin, and B. W. J. H. Penninx, “Longitudinal Associations Between Metabolic Syndrome Components and Telomere Shortening,” J Clin Endocrinol Metab, vol. 100, no. 8, pp. 3050–3059, Aug. 2015
[16] O. Ramos-Lopez, J. I. Riezu-Boj, and F. I. Milagro, “Genetic and epigenetic nutritional interactions influencing obesity risk and adiposity outcomes,” Curr Opin Clin Nutr Metab Care, vol. 25, no. 4, pp. 235–240, Jul. 2022
[17] P. Cordero, J. Li, and J. A. Oben, “Epigenetics of obesity: Beyond the genome sequence,” Curr Opin Clin Nutr Metab Care, vol. 18, no. 4, pp. 361–366, Jul. 2015
[18] R. S. Strakovsky, X. Zhang, D. Zhou, and Y. X. Pan, “Gestational high fat diet programs hepatic phosphoenolpyruvate carboxykinase gene expression and histone modification in neonatal offspring rats,” J Physiol, vol. 589, no. Pt 11, pp. 2707–2717, Jun. 2011
[19] F. Ottens, A. Franz, and T. Hoppe, “Build-UPS and break-downs: metabolism impacts on proteostasis and aging,” Cell Death Differ, vol. 28, no. 2, p. 505, Feb. 2021
[20] U. Özcan et al., “Endoplasmic reticulum stress links obesity, insulin action, and type 2 diabetes,” Science, vol. 306, no. 5695, pp. 457–461, Oct. 2004
[21] B. Lu and S. Guo, “Mechanisms Linking Mitochondrial Dysfunction and Proteostasis Failure,” Trends Cell Biol, vol. 30, no. 4, pp. 317–328, Apr. 2020
[22] G. S. Hotamisligil, “Endoplasmic reticulum stress and the inflammatory basis of metabolic disease,” Cell, vol. 140, no. 6, pp. 900–917, 2010
[23] N. Gregersen and P. Bross, “Protein misfolding and cellular stress: An overview,” Methods in Molecular Biology, vol. 648, pp. 3–23, 2010
[24] S. Z. Hasnain, R. Lourie, I. Das, A. C. H. Chen, and M. A. McGuckin, “The interplay between endoplasmic reticulum stress and inflammation,” Immunol Cell Biol, vol. 90, no. 3, pp. 260–270, Mar. 2012
[25] H. A. James, B. T. O’Neill, and K. S. Nair, “Insulin Regulation of Proteostasis and Clinical Implications,” Cell Metab, vol. 26, no. 2, p. 310, Aug. 2017
[26] N. Ho, C. Xu, and G. Thibault, “From the unfolded protein response to metabolic diseases – Lipids under the spotlight,” J Cell Sci, vol. 131, no. 3, Feb. 2018
[27] J. Chaudhuri et al., “The role of advanced glycation end products in aging and metabolic diseases: bridging association and causality,” Cell Metab, vol. 28, no. 3, p. 337, Sep. 2018
[28] N. Mizushima, T. Yoshimori, and Y. Ohsumi, “The Role of Atg Proteins in Autophagosome Formation,” Annual review of cell and developmental biology, vol. 27, pp. 107–132, Oct. 2011
[29] R. Singh et al., “Autophagy regulates lipid metabolism,” Nature, vol. 458, no. 7242, pp. 1131–1135, Apr. 2009
[30] G. Ashrafi and T. L. Schwarz, “The pathways of mitophagy for quality control and clearance of mitochondria,” Cell Death Differ., vol. 20, no. 1, pp. 31–42, Jan. 2013
[31] C. López-Otín, M. A. Blasco, L. Partridge, M. Serrano, and G. Kroemer, “Hallmarks of aging: An expanding universe,” Cell, vol. 186, no. 2, pp. 243–278, Jan. 2023.
[32] M. Laplante and D. M. Sabatini, “mTOR signaling in growth control and disease,” Cell, vol. 149, no. 2, pp. 274–293, Apr. 2012
[33] V. T. Samuel and G. I. Shulman, “Mechanisms for insulin resistance: Common threads and missing links,” Cell, vol. 148, no. 5, pp. 852–871, Mar. 2012
[34] V. T. Samuel and G. I. Shulman, “The pathogenesis of insulin resistance: integrating signaling pathways and substrate flux,” J Clin Invest, vol. 126, no. 1, pp. 12–22, Jan. 2016
[35] C. B. Newgard, “Interplay between lipids and branched-chain amino acids in development of insulin resistance,” Cell Metab, vol. 15, no. 5, pp. 606–614, May 2012
[36] R. Stinkens, G. H. Goossens, J. W. E. Jocken, and E. E. Blaak, “Targeting fatty acid metabolism to improve glucose metabolism,” Obesity Reviews, vol. 16, no. 9, pp. 715–757, Sep. 2015
[37] R. C. R. Meex, E. E. Blaak, and L. J. C. van Loon, “Lipotoxicity plays a key role in the development of both insulin resistance and muscle atrophy in patients with type 2 diabetes,” Obes Rev, vol. 20, no. 9, pp. 1205–1217, 2019
[38] G. S. Hotamisligil, “Inflammation and metabolic disorders,” Nature 2006 444:7121, vol. 444, no. 7121, pp. 860–867, Dec. 2006
[39] Y. Alwarawrah, K. Kiernan, and N. J. MacIver, “Changes in nutritional status impact immune cell metabolism and function,” Front Immunol, vol. 9, no. MAY, p. 1055, May 2018
[40] B. Khambu, S. Yan, N. Huda, G. Liu, and X. M. Yin, “Autophagy in non-alcoholic fatty liver disease and alcoholic liver disease,” Liver Res, vol. 2, no. 3, pp. 112–119, Sep. 2018
[41] C. López-Otín, M. A. Blasco, L. Partridge, M. Serrano, and G. Kroemer, “The hallmarks of aging,” Cell, vol. 153, no. 6, p. 1194, Jun. 2013.
[42] K. Kolczynska, A. Loza-Valdes, I. Hawro, and G. Sumara, “Diacylglycerol-evoked activation of PKC and PKD isoforms in regulation of glucose and lipid metabolism: a review,” Lipids in Health and Disease 2020 19:1, vol. 19, no. 1, pp. 1–15, May 2020
[43] R. J. Mailloux, “An Update on Mitochondrial Reactive Oxygen Species Production,” Antioxidants 2020, Vol. 9, Page 472, vol. 9, no. 6, p. 472, Jun. 2020
[44] M. J. López-Armada, R. R. Riveiro-Naveira, C. Vaamonde-García, and M. N. Valcárcel-Ares, “Mitochondrial dysfunction and the inflammatory response,” Mitochondrion, vol. 13, no. 2, pp. 106–118, Mar. 2013
[45] D. Muñoz-Espín and M. Serrano, “Cellular senescence: From physiology to pathology,” Nat Rev Mol Cell Biol, vol. 15, no. 7, pp. 482–496, 2014
[46] R. Spinelli et al., “Increased cell senescence in human metabolic disorders,” J Clin Invest, vol. 133, no. 12, 2023
[47] M. Monserrat-Mesquida et al., “Metabolic Syndrome Is Associated with Oxidative Stress and Proinflammatory State,” Antioxidants 2020, Vol. 9, Page 236, vol. 9, no. 3, p. 236, Mar. 2020
[48] D. Révész, Y. Milaneschi, J. E. Verhoeven, J. Lin, and B. W. J. H. Penninx, “Longitudinal Associations Between Metabolic Syndrome Components and Telomere Shortening,” J Clin Endocrinol Metab, vol. 100, no. 8, pp. 3050–3059, Aug. 2015
[49] N. Gregersen and P. Bross, “Protein misfolding and cellular stress: An overview,” Methods in Molecular Biology, vol. 648, pp. 3–23, 2010
[50] K. S. Rajagopalan et al., “Metabolic Syndrome Induces Epigenetic Alterations in Mitochondria-Related Genes in Swine Mesenchymal Stem Cells,” Cells, vol. 12, no. 9, p. 1274, May 2023
[51] E. Mansilla et al., “Could metabolic syndrome, lipodystrophy, and aging be mesenchymal stem cell exhaustion syndromes?,” Stem Cells Int, 2011
[52] R. Soares, “Angiogenesis in the metabolic syndrome,” Oxidative Stress, Inflammation and Angiogenesis in the Metabolic Syndrome, pp. 85–99, 2009
[53] A. Salminen, K. Kaarniranta, and A. Kauppinen, “Insulin/IGF-1 signaling promotes immunosuppression via the STAT3 pathway: impact on the aging process and age-related diseases,” Inflammation Research, vol. 70, no. 10–12, pp. 1043–1061, 2021
[54] S. Hurrle and W. H. Hsu, “The etiology of oxidative stress in insulin resistance,” Biomed J, vol. 40, no. 5, pp. 257–262, Oct. 2017
[55] K. Esposito and D. Giugliano, “The metabolic syndrome and inflammation: association or causation?,” Nutrition, Metabolism and Cardiovascular Diseases, vol. 14, no. 5, pp. 228–232, Oct. 2004
[56] M. Kabátková et al., “Interactive effects of inflammatory cytokine and abundant low-molecular-weight PAHs on inhibition of gap junctional intercellular communication, disruption of cell proliferation control, and the AhR-dependent transcription,” Toxicol Lett, vol. 232, no. 1, pp. 113–121, Jan. 2015
[57] B. B. BRANDAO, E. ALTINDIS, R. GARCIA MARTIN, and C. R. KAHN, “Serum Exosomal Proteins—A New Component of Intercellular Communication in Metabolism,” Diabetes, vol. 67, no. Supplement_1, Jul. 2018
[58] P. Trayhurn, B. Wang, and I. S. Wood, “Hypoxia in adipose tissue: a basis for the dysregulation of tissue function in obesity?,” Br J Nutr, vol. 100, no. 2, pp. 227–235, 2008
[59] P. X. Wang, X. R. Deng, C. H. Zhang, and H. J. Yuan, “Gut microbiota and metabolic syndrome,” Chin Med J (Engl), vol. 133, no. 7, p. 808, Apr. 2020
[60] M. Arrese, D. Cabrera, A. M. Kalergis, and A. E. Feldstein, “Innate Immunity and Inflammation in NAFLD/NASH,” Dig Dis Sci, vol. 61, no. 5, pp. 1294–1303, May 2016
[61] M. M. Yeh and E. M. Brunt, “Pathological features of fatty liver disease,” Gastroenterology, vol. 147, no. 4, pp. 754–764, Oct. 2014
[62] V. Guarner and M. E. Rubio-Ruiz, “Low-Grade Systemic Inflammation Connects Aging, Metabolic Syndrome and Cardiovascular Disease,” Interdiscip Top Gerontol, vol. 40, pp. 99–106, Oct. 2014
[63] T. Caputo, F. Gilardi, and B. Desvergne, “From chronic overnutrition to metaflammation and insulin resistance: adipose tissue and liver contributions,” FEBS Lett, vol. 591, no. 19, pp. 3061–3088, Oct. 2017
[64] Q. Yuan, Z. L. Zeng, S. Yang, A. Li, X. Zu, and J. Liu, “Mitochondrial Stress in Metabolic Inflammation: Modest Benefits and Full Losses,” Oxid Med Cell Longev, vol. 2022, 2022
[65] K. Rabe, M. Lehrke, K. G. Parhofer, and U. C. Broedl, “Adipokines and Insulin Resistance,” Molecular Medicine 2008 14:11, vol. 14, no. 11, pp. 741–751, Nov. 2008
[66] J. Grootjans, A. Kaser, R. J. Kaufman, and R. S. Blumberg, “The unfolded protein response in immunity and inflammation,” Nature Reviews Immunology 2016 16:8, vol. 16, no. 8, pp. 469–484, Jun. 2016
[67] S. Carding, K. Verbeke, D. T. Vipond, B. M. Corfe, and L. J. Owen, “Dysbiosis of the gut microbiota in disease,” Microb Ecol Health Dis, vol. 26, no. 0, Feb. 2015
[68] E. Amabebe, F. O. Robert, T. Agbalalah, and E. S. F. Orubu, “Microbial dysbiosis-induced obesity: role of gut microbiota in homoeostasis of energy metabolism,” British Journal of Nutrition, vol. 123, no. 10, pp. 1127–1137, May 2020
[69] E. Lazar, A. Sherzai, J. Adeghate, and D. Sherzai, “Gut dysbiosis, insulin resistance and Alzheimer’s disease: Review of a novel approach to neurodegeneration,” Frontiers in Bioscience – Scholar, vol. 13, no. 1, pp. 17–29, Jun. 2021
[70] M. V. Salguero, M. A. I. Al-Obaide, R. Singh, T. Siepmann, and T. L. Vasylyeva, “Dysbiosis of Gram-negative gut microbiota and the associated serum lipopolysaccharide exacerbates inflammation in type 2 diabetic patients with chronic kidney disease,” Exp Ther Med, vol. 18, no. 5, pp. 3461–3469, Nov. 2019
[71] R. Tyszkowski and R. Mehrzad, “Inflammation: A multifaceted and omnipresent phenomenon,” Inflammation and Obesity: A New and Novel Approach to Manage Obesity and its Consequences, pp. 19–30, Jan. 2023
[72] C. O. M. Jasirwan, C. R. A. Lesmana, I. Hasan, A. S. Sulaiman, and R. A. Gani, “The role of gut microbiota in non-alcoholic fatty liver disease: pathways of mechanisms,” Biosci Microbiota Food Health, vol. 38, no. 3, p. 81, 2019
[73] J. Myers, P. Kokkinos, and E. Nyelin, “Physical Activity, Cardiorespiratory Fitness, and the Metabolic Syndrome,” Nutrients 2019, Vol. 11, Page 1652, vol. 11, no. 7, p. 1652, Jul. 2019
[74] I. Hoyas and M. Leon-Sanz, “Nutritional Challenges in Metabolic Syndrome,” Journal of Clinical Medicine 2019, Vol. 8, Page 1301, vol. 8, no. 9, p. 1301, Aug. 2019
[75] S. M. Grundy et al., “Diagnosis and management of the metabolic syndrome: An American Heart Association/National Heart, Lung, and Blood Institute scientific statement,” Circulation, vol. 112, no. 17, pp. 2735–2752, Oct. 2005
[76] J. Wärnberg, S. Gomez-Martinez, J. Romeo, L. E. Díaz, and A. Marcos, “Nutrition, Inflammation, and Cognitive Function,” Ann N Y Acad Sci, vol. 1153, no. 1, pp. 164–175, Feb. 2009
[77] Z. Lv and Y. Guo, “Metformin and Its Benefits for Various Diseases,” Front Endocrinol (Lausanne), vol. 11, p. 191, Apr. 2020
[78] T. Pham et al., “MitoQ and CoQ10 supplementation mildly suppresses skeletal muscle mitochondrial hydrogen peroxide levels without impacting mitochondrial function in middle-aged men,” European Journal of Applied Physiology 2020 120:7, vol. 120, no. 7, pp. 1657–1669, May 2020
[79] E. O. Wissler Gerdes, Y. Zhu, T. Tchkonia, and J. L. Kirkland, “Discovery, development, and future application of senolytics: theories and predictions,” FEBS J, vol. 287, no. 12, pp. 2418–2427, Jun. 2020
[80] R. Kumar, U. Sood, V. Gupta, M. Singh, J. Scaria, and R. Lal, “Recent Advancements in the Development of Modern Probiotics for Restoring Human Gut Microbiome Dysbiosis,” Indian Journal of Microbiology 2019 60:1, vol. 60, no. 1, pp. 12–25, May 2019
[81] W. Zakrzewski, M. Dobrzynski, M. Szymonowicz, and Z. Rybak, “Stem cells: Past, present, and future,” Stem Cell Res Ther, vol. 10, no. 1, pp. 1–22, Feb. 2019



